


In recent years, oncology drug developers have invested in antibody-drug conjugates (ADCs). These targeted therapies deliver cytotoxic small-molecule drug payloads to cancer cells. The cytotoxic payload attaches to a monoclonal antibody (designed for binding to the target) through a linker.
Having a solid clinical pharmacology strategy is essential to successfully developing these therapies. The US Food and Drug Administration (FDA) issued the draft Clinical Pharmacology Considerations for Antibody-Drug Conjugates (ADC), Guidance for Industry in February 2022. The agency finalized the guidance in March 2024.
While the revision remains largely unchanged, it includes a few updates. One example is the addition of assessing both fixed and weight-based dosing strategies. The DDI section has been condensed.
The guidance provides a framework for ADC drug developers, particularly for the payload. The guidance covers many areas central to clinical pharmacology including:
- bioanalytical methods,
- dosing strategies,
- dose- and exposure-response analysis,
- intrinsic patient factors,
- QTc assessments of proarrhythmic potential,
- immunogenicity,
- pharmacogenomics, and
- drug-drug interactions (DDIs).
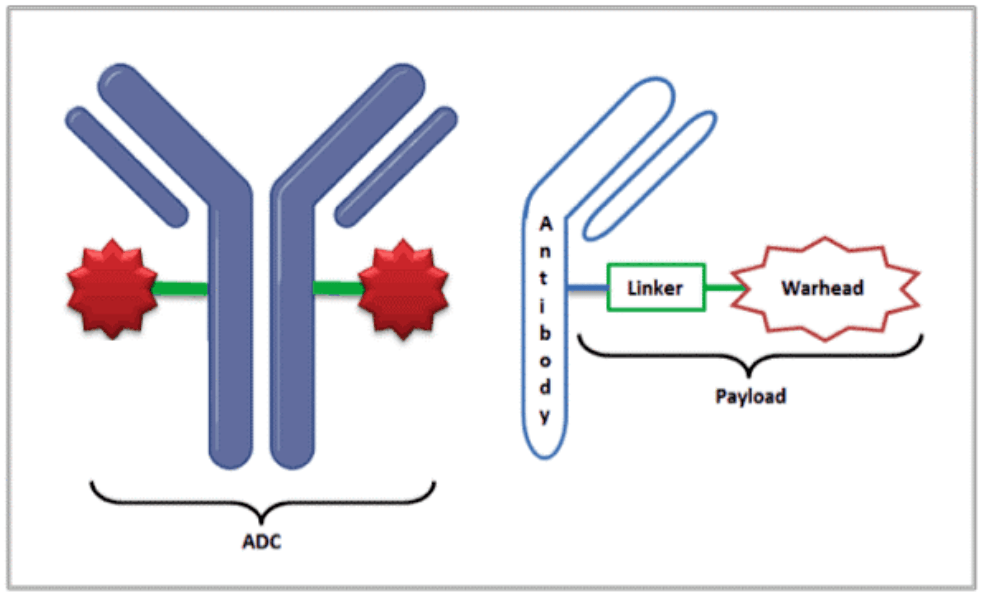
The guidance shares insights on specific ADC analytic moieties, including free and total antibody, free small molecule, linker, and pharmacologically active small molecule metabolites. It acknowledges the challenges related to dose adjustments based on patient intrinsic and extrinsic factors. Many topics are consistent with the agency’s feedback on individual programs and/or existing clinical pharmacology guidance.
Here are a few key areas that may interest you.
Bioanalysis
The guidance is specific to analytes that sponsors should consider measuring depending on the drug development phase. For example, measure the ADC, its constituents, and its pharmacologically active metabolites in first-in-human (FIH) studies. Later in development, the ADC, its constituent, and its pharmacologically active metabolites that are quantifiable in systemic circulation should be measured to inform exposure-response analyses.
The guidance also provides provisions to exclude measuring ADC constituents or pharmacologically active metabolites in later development. Interestingly, the guidance provides some specificity on the systemic presence of the shed target. If significant, the bioanalytical assays need to distinguish the target-unbound from target-bound ADC.
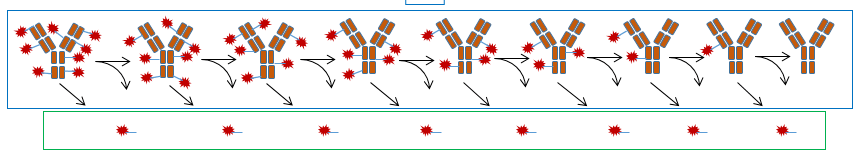
Clinical ADC characterization sometimes hinges on the presence of multiple molecular species, including the different drug-to-antibody (DAR) species. The changing nature of the mixture in vivo and various catabolites/adducts can complicate this analysis. Developing quantitative bioanalytical assays is challenging because different DAR species might behave differently in the assays. It’s unclear whether the metabolites in safety testing (MIST) guidance will apply to pharmacologically active metabolites (a metabolite with pharmacological activity at the target receptor).
Exposure/response analysis
To support dose selection, conduct exposure-response analyses of clinical trials for safety and efficacy with the ADC, its constituents, and any pharmacologically active metabolites. In later development, justification could be provided for not conducting exposure-response analyses with an ADC constituent or pharmacologically active metabolites. If the antibody target is shed significantly into the systemic circulation, exposure-response analyses should only be conducted with the ADC and/or total antibody that is unbound to the circulating shed target.
Developing E/R models for ADCs presents several challenges. These include the presence of multiple analytes for ADCs with physiochemical characteristics that may impact disposition, the changing nature of the ADC regarding DARs in vivo, as well as potential immunogenicity that can cause anti-drug antibody (ADA) formation.
Intrinsic factors in the patient population of interest
The guidance suggests that intrinsic factors (e.g., renal or liver impairment, pharmacogenomics, body weight, age, gender, race) that can influence exposure of the ADC, its constituent parts, and pharmacologically active metabolites, if any, should be evaluated in either: 1) clinical studies, through population pharmacokinetic (PK) analysis; or 2) dedicated studies.
This is consistent with other FDA guidances on this subject. Notably, a population pharmacokinetic approach can be used to assess the effects of organ impairment on the unconjugated payload, pharmacologically active metabolites, if any, and/or other ADC constituent parts if patients with organ impairment are enrolled in pivotal studies, and pharmacokinetic data coupled with safety and efficacy information in those patients are available. Such an approach increases the development program’s probability of success.
QTc assessment for cardiac safety
The unconjugated payload is the only part of the ADC with potential risk for QT prolongation (Figure 3). Any ADC QT assessment plan should consider all the factors in a small-molecule drug QT assessment.

The ADC is unlikely to interact with the human Ether-à-go-go-Related Gene (hERG) channel because of the low concentrations of circulating payload after ADC dosing. In general, a cQT analysis showing no meaningful effect should support a TQT study waiver. When time-matched PK and QT samples aren’t collected, the ICH E14 guideline allows using modeled concentrations.
Don’t take this aspect lightly! The agency can request clinical assessment of QT prolongation risk. Studies were done to evaluate the risk of QTc prolongation in patients with CD30-positive hematologic malignancies (for brentuximab-vedontin) and HER2-positive metastatic breast cancer patients (for trastuzumab emtansine).
Drug-drug interaction risk assessment
ADC development programs should assess in vitro DDI risk for the unconjugated payload and pharmacologically active metabolites, if any, as both a perpetrator and a victim using both CYP enzyme- and transporter-related assays. The FDA could recommend that the sponsor conduct an in vivo DDI evaluation of the unconjugated payload as a victim and consider physiologically-based pharmacokinetic (PBPK) modeling for characterizing the probability of an effect. The FDA could also recommend assessing the DDI potential for the antibody component.
In general, extensive in vitro DDI risk assessment helps rationalize clinical DDI study waivers. The FDA supports using PBPK modeling to strengthen the evidence. The agency has borrowed drug interaction information from other approved ADCs. A sponsor taking such an approach may be limited to situations with right of reference.
Dosing to maximize the therapeutic index
The evolution of the FDA’s Project Optimus and the new guidance means that dose optimization principles also apply to ADCs. Because ADCs have both biologic and small molecule components, these aspects can be tricky.
One recent example is Teliso-V (Telisotuzumab vedotin) an ADC composed of the anti–c-Met humanized monoclonal antibody ABT-700 coupled to the cytotoxic monomethyl auristatin E (MMAE) through a valine–citrulline linker (ABT-700–vcMMAE). Incidentally, the FDA–approved ADC, brentuximab vedotin uses the same linker–drug payload.
Teliso-V delivers MMAE specifically to c-Met–expressing tumor cells. Engagement of c-Met by Teliso-V results in ADC internalization and intracellular release of MMAE after linker proteolysis. MMAE then binds to tubulin, thereby inhibiting mitosis and killing tumor cells.
Abbvie recently tweaked telisotuzumab vedotin dose from 2.7 mg/kg Q3W as the RP2D (recommended phase 2 dose) to 1.9 mg/kg Q2W (Camidge et al., 2021). These adjustments were a result of designing the right experiment and applying exposure/response relationships in determining the optimal dose.
In summary, little is new in the latest ADC clinical pharmacology guidance. However, this guidance will help sponsors pay attention to several considerations including:
- In vitro and in vivo ADME characterization during preclinical development, including identification of metabolites and determining their pharmacological activity
- Bioanalytical method strategy development before clinical evaluation
- Translational PK/PD strategy for payload and linker components
- Dose and regimen selection during early clinical development using exposure/response
- Concentration/QT assessment within the FIH study
- Modeling and simulation strategy that includes developing population PK, PK/PD, and PBPK models to interrogate the effects of intrinsic and extrinsic factors
For an example of how modeling and simulation supported an ADC development program, watch this webinar.
References
Camidge DR et al. Phase I Study of 2- or 3-Week Dosing of Telisotuzumab Vedotin, an Antibody–Drug Conjugate Targeting c-Met, Monotherapy in Patients with Advanced Non–Small Cell Lung Carcinoma. Clin Cancer Res; 27(21) November 1, 2021
Krishna, R., 2021. Key Considerations to Ensure Maximal Probability of Antibody Drug Conjugate Development Success. [Blog] Certara [Accessed 18 April 2022].
Sikorski, S.R.I.Q.R. The Clinical Landscape of Antibody-drug Conjugates. 2014 [cited 2021 30 September];
This blog was originally published by April 21, 2022 and has since been updated May 10, 2024.
